Статьи
Эпилептические энцефалопатии
Наиболее тяжелые формы эпилепсии приходятся на ранний период жизни, представляя большое испытание для эпилептологов, неврологов и педиатров. При изучении эпилепсии детского возраста особое внимание уделяется энцефалопатиям развития и эпилептическим энцефалопатиям, сложным не только в вопросе постановки диагноза, но и в тактике лечения.
ТЯЖЕЛЫЕ ЭПИЛЕПСИИ У ДЕТЕЙ
С эпилепсией — одним из наиболее распространенных неврологических заболеваний — сталкиваются до 1 % людей. Болезнь проявляется избыточной активностью головного мозга с кратковременным нарушением его функции. Не все виды эпилепсии протекают одинаково тяжело. Около 70 % пациентов удается подобрать эффективную терапию. Самые трудные случаи встречаются у детей раннего возраста, о которых и пойдет речь.
В 2017 году была опубликована «Классификация эпилепсий Международной противоэпилептической лиги (МПЭЛ)» с изложением позиции комиссии МПЭЛ по классификации и терминологии. Целевая группа создала отдельную категорию для синдромов, связанных с энцефалопатией развития и (или) эпилептической энцефалопатией (ЭРЭ), или синдромов с прогрессирующим неврологическим дефицитом.
Точная распространенность ЭРЭ в РФ неизвестна, в отдельных литературных источниках доля эпилептических энцефалопатий составляет 7–15 % от всех форм эпилепсии, достигая почти 40 % среди возникающих в первые 3 года жизни. Некоторые формы встречаются более часто, например синдром Драве (заболеваемость которым составляет от 0,5–1,0 случая на 40 тыс. до 1,0 на 15,7 тыс.) или STXBP1-ЭРЭ с прогнозируемой частотой 3,30–3,81 на 100 тыс. рождений.
Ядро клинических проявлений ЭРЭ представляет собой сочетание разнообразных эпилептических приступов и нарушения развития как в виде прогрессирующего ухудшения (регресса развития), так и статичной задержки (так называемого плато развития) психического, моторного и речевого развития.
При этом считается, что к задержке развития ребенка приводит как непосредственно первопричина болезни (ДЦП, генетическая этиология и т.п.), так и агрессивное течение самой эпилепсии.
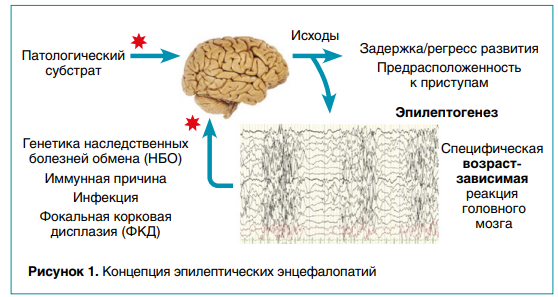
ЭРЭ могут возникать в любом возрасте, но чаще всего встречаются у младенцев и детей до трех лет, что объясняется особенностями нейрогенеза растущего мозга. Такое негативное влияние патологической активности на электроэнцефалограмме (ЭЭГ) и приступов в критические периоды развития головного мозга не только ухудшает уже существующие функции, но и затормаживает возрастное развитие новых (рис. 1).
ОСОБЕННОСТИ ТЕЧЕНИЯ ЭРЭ
Термин «эпилептическая энцефалопатия» не означает нарушение развития с рождения. Нередко началу эпилепсии и прогрессирующим неврологическим симптомам может предшествовать совершенно нормальное раннее развитие ребенка. Важно на самых ранних этапах заметить, что мы имеем дело с эпилептическими энцефалопатиями, так как своевременно начатое лечение обеспечивает лучшие исходы в отношении развития и качества жизни.
Эпилептические приступы при ЭРЭ крайне разнообразны: короткие вздрагивания (миоклонические приступы), «складывания» или приподнимание рук (эпилептические спазмы), напряжение (тонические приступы) или ритмичные подергивания (клонические пароксизмы) всего тела либо одной конечности, замирания (абсансы) и др. Нередко можно встретить сразу несколько типов приступов (чаще всего инфантильные спазмы и тонические приступы) у одного пациента или же трансформацию одних типов приступов в другие с течением времени, что требует регулярного наблюдения за больными.
Эпилептические энцефалопатии сопряжены с высоким риском синдрома внезапной смерти при эпилепсии (SUDEP), что ставит задачу контроля над приступами на первое место. Особую опасность для жизни и здоровья представляют эпилептические статусы, когда приступ не заканчивается самостоятельно и длится дольше 5 минут, что требует надлежащего лечения в как можно более короткие сроки.
Долгосрочный исход ЭРЭ связан как с этиологическими факторами (поиск ответа на вопрос, почему это вообще началось), так и с факторами, связанными с эпилепсией (насколько тяжело протекает болезнь),— возрастом начала эпилепсии, типами приступов, видом электроэнцефалографических нарушений, продолжительностью заболевания. В большинстве случаев прогноз неблагоприятный из-за высокого уровня инвалидизации, повышенного риска SUDEP, по сравнению с другими формами, и фармакорезистентностью.
Тем не менее при некоторых этиологиях и подборе оптимального лечения можно добиться стабилизации болезни и почти нормального уровня интеллекта при долгосрочном наблюдении в более чем 20 % случаев.
В ПОИСКАХ ПЕРВОПРИЧИНЫ
Вышеперечисленные симптомы могут проявляться практически в любой комбинации — в разном возрасте, с разными типами приступов, с нормальной или грубо измененной ЭЭГ в дебюте болезни, что значительно осложняет постановку четкого диагноза и прогнозирование ее течения. В диагностике широко используются методы ЭЭГ, оценки феноменологии (как выглядит приступ), нейровизуализация с помощью магнитно-резонансной и компьютерной томографии (МРТ и КТ) головного мозга, генетические и метаболические тесты.
Этиология эпилепсий весьма разнообразна и классифицируется по пяти основным направлениям: структурные, генетические, инфекционные, метаболические и иммунные. Однако международный консенсус подчеркивает особый вклад генетических изменений в возникновение ЭРЭ. Подтверждение молекулярно-генетического диагноза позволяет не только установить точную причину болезни и ожидаемый прогноз, но и не проводить ненужное дополнительное обследование, а в ряде случае даже подобрать более эффективное лечение и провести профилактику заболевания в семье.
Концепция генетических или наследственных эпилепсий не определяется исключительно передачей заболевания внутри одной семьи, а основывается на нарушении нормальной структуры наследственного материала (ДНК) в области генов или путем эпигенетического механизма. Другими словами, нарушается «письменная» инструкция, как работать клетке, и это приводит к развитию симптомов болезни.
Такие нарушения могут возникать de novo (спонтанно, без передачи от родителей) и наследоваться (например, Х-сцепленный или рецессивный тип наследования), что делает постановку молекулярногенетического диагноза крайне важным: риски повторения болезни в семье могут достигать 25–50 %! К счастью, сегодня в медицинском арсенале имеются доступные методы профилактики наследственной эпилепсии, а именно — предимплантационное или пренатальное тестирование при планировании следующих беременностей.
ГЕНЕТИЧЕСКАЯ ДИАГНОСТИКА
Выявляемость генетических причин варьирует на уровне 30–60 %. Выявляемости на 100 % не позволяют достичь биологические и технические ограничения: до сих пор связь с болезнями хорошо изучена не у всех генов, базы данных о патогенных или доброкачественных вариантах нуклеотидной последовательности все время пополняются, меняются методы обработки информации, что требует постоянного обновления подхода к диагностике и пересмотра данных уже проведенных исследований каждые полтора-два года.
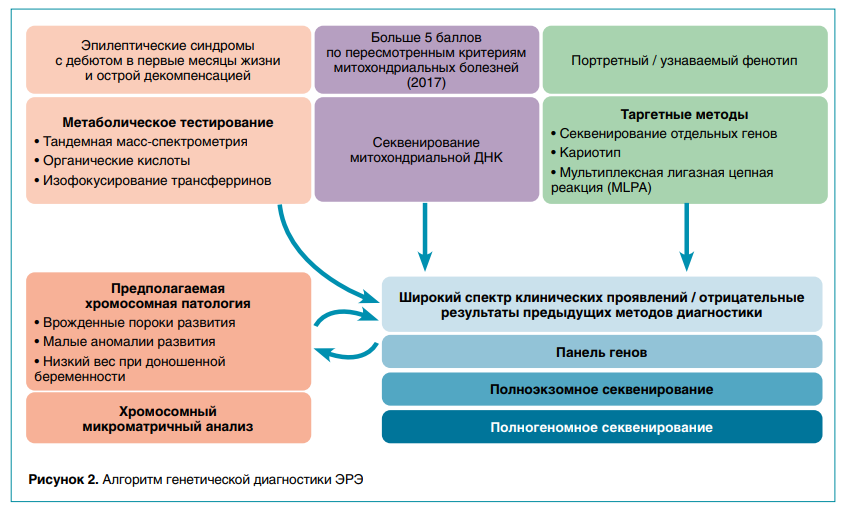
Можно сформировать некоторые общие рекомендации по диагностическому алгоритму: при узнаваемом (портретном) фенотипе имеет смысл стартовать с узконаправленных методов (секвенирование отдельных генов, кариотип, мультиплексная лигазная цепная реакция, MLPA); при подозрении на болезни обмена полезно проведение метаболического тестирования (тандемная масс-спектрометрия, органические кислоты, изофокусирование трансферринов и др.), особенно при эпилептических синдромах с дебютом в первые месяцы жизни и с острой декомпенсацией.
При сочетании ЭРЭ с врожденными пороками и малыми аномалиями развития диагностику следует начинать с хромосомного микроматричного анализа (ХМА), в процессе которого исследуют все клинически значимые участки генома и определяют избыток или недостаток генетического материала (числовые аномалии, дупликации и делеции части хромосом).
Однако наиболее эффективной стратегией считается использование высокопроизводительного секвенирования (англ. next generation sequencing, NGS), которое подразделяется на две методологически разные группы:
• экзомное секвенирование, когда исследуются белок-кодирующие участки ДНК (экзоны);
• геномное секвенирование, когда равномерно покрывается практически вся ДНК, что позволяет хорошо выявлять большее количество уникальных вариантов как в экзонах, так и в интронах, анализировать митохондриальную ДНК и возможные хромосомные перестройки и тандемные повторы.
Современный диагностический алгоритм (рис. 2) не выделяет какой-либо из вышеперечисленных тестов как единственный и всеобъемлющий метод обследования. При отрицательном результате в одном из исследований предлагается переходить к следующему из неиспользованных тестов. Верхушкой диагностического алгоритма является полногеномное секвенирование трио, когда обследование проводится в «ядерной семье» (пробанд и его биологические родители).
ОТДЕЛЬНЫЕ ГРУППЫ ЭРЭ
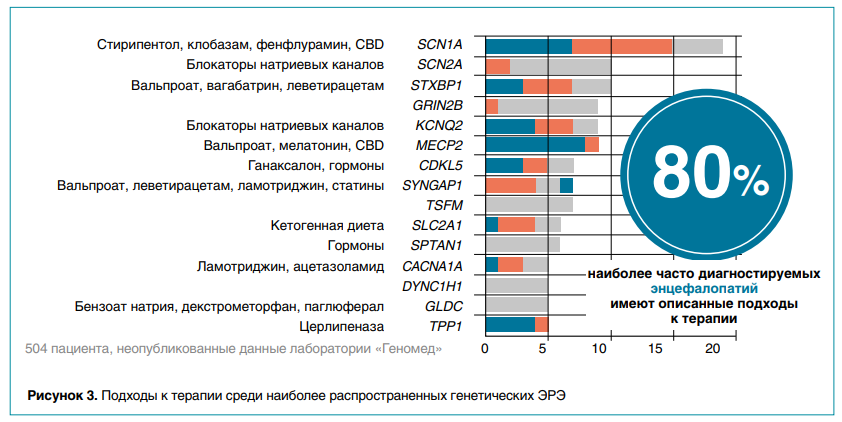
Около 80 % наиболее часто диагностируемых генетических эпилептических энцефалопатий требуют подходов к лечению, описанных на рисунке 3. Сегодня в терапии ЭРЭ используют весьма агрессивную тактику с быстрым перебором противосудорожных препаратов и использованием высокодозной гормональной терапии или внутривенных иммуноглобулинов либо специальной кетогенной диеты. Также в некоторых случаях рассматривают хирургическое лечение, например установку стимулятора блуждающего нерва (VNS) или рассечение мозолистого тела (каллозотомия).
За последние 10 лет клинический опыт отдела психоневрологии и эпилептологии НИКИ педиатрии и детской хирургии имени Ю.Е. Вельтищева позволил нам выделить следующие группы ЭРЭ с наиболее эффективными подходами к терапии:
1) для SCN2A/SCN8A-ЭРЭ ожидается эффективность блокаторов натриевых каналов, которые не следует использовать при SCN1A-ЭРЭ из-за значительного риска провокации эпилептических приступов;
2) при TSC1/2—туберозном склерозе препаратом выбора является вигабатрин;
3) SCN1A-ЭРЭ зачастую требуют использования ранней комбинированной противосудорожной терапии, наиболее эффективными из доступных в РФ считаются препараты вальпроевой кислоты и клобазам. В качестве дополнительных опций лечения следует рассматривать кетогенную диету и имплантацию стимулятора блуждающего нерва;
4) STXBP1-ЭРЭ положительно реагируют на препараты вальпроевой кислоты, леветирацетам и вигабатрин;
5) при CDKL5-ЭРЭ показал эффективность препарат ганаксолон, который исследовался в институте имени Ю.Е. Вельтищева, но пока не прошел окончательную регистрацию и недоступен всем пациентам в РФ;
6) при SLC2A1-ЭРЭ следует использовать кетогенную диету в первой линии как патогенетическое лечение.
Для других ЭРЭ со специфичной этиологией оптимальные подходы к терапии только изучаются, хотя в литературе можно найти одиночные описания эффективности того или иного препарата.
ПЕРСПЕКТИВЫ ЛЕЧЕНИЯ
Кроме отдельных противосудорожных препаратов, активно разрабатываются методики генной терапии, прямо или опосредованно направленные на изменение функции белкового продукта гена. Такие разработки—ресурсоемкие, требуют привлечения большого числа специалистов из смежных областей и нерентабельны, поскольку пациентов, которым необходимо подобное лечение, мало. Это требует отдельного внимания со стороны государства и научных коллективов. Важность данного направления можно проиллюстрировать историей разработки терапии для спинальной мышечной атрофии (СМА), изменившей течение и исходы заболевания при своевременно начатом лечении. Возможность кардинальным образом модифицировать болезнь с помощью генной терапии позволит не только уменьшить нагрузку на систему здравоохранения за счет снижения инвалидизации, но и повлиять на здоровье нации в долгосрочной перспективе.
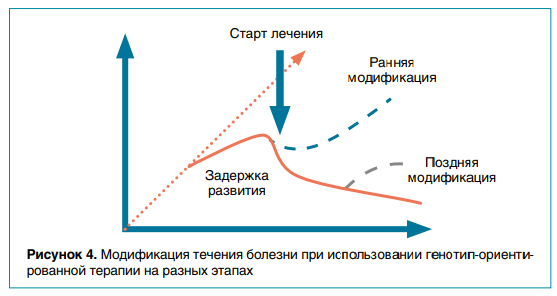 В мире есть несколько удачно завершившихся примеров патогенетического/этиологического лечения: миласен для нейронального цероидного липофусциноза (НЦЛ) 7-го типа и Бринейра для НЦЛ 2-го типа. Сейчас гораздо больше терапевтических опций находится на этапе разработки или ввода в клинические испытания (например, антисмысловые олигонуклеотиды для синдрома Драве). Вот почему крайне важна как можно более ранняя диагностика этиологии ЭРЭ для начала своевременной терапии и улучшения течения болезни (рис. 4). Подобные исследования, как международные, так и инициативные, проводятся в России на базе института имени Ю.Е. Вельтищева
В мире есть несколько удачно завершившихся примеров патогенетического/этиологического лечения: миласен для нейронального цероидного липофусциноза (НЦЛ) 7-го типа и Бринейра для НЦЛ 2-го типа. Сейчас гораздо больше терапевтических опций находится на этапе разработки или ввода в клинические испытания (например, антисмысловые олигонуклеотиды для синдрома Драве). Вот почему крайне важна как можно более ранняя диагностика этиологии ЭРЭ для начала своевременной терапии и улучшения течения болезни (рис. 4). Подобные исследования, как международные, так и инициативные, проводятся в России на базе института имени Ю.Е. Вельтищева
Читайте также
- Цифровой фенотип: современная информационно-аналитическая платформа для выявления новых гено-фенотипических связей
- История становления отечественной детской нейроурологии
- К истокам отечественной пульмонологии детского возраста: к 100-летию профессора С.Ю. Каганова
- Как правильно написать научную медицинскую статью
НАШИ ПАРТНЕРЫ