Конец диагностической одиссеи. Как клиническая геномика меняет мышление врача-генетика
В последние десятилетия мы стали свидетелями и участниками технологической революции, драматическим образом повлиявшей на деятельность врачей-генетиков. Появление хромосомного микроматричного анализа и высокопроизводительного секвенирования ДНК предоставило нам беспрецедентные по своим возможностям методы генетического тестирования, которые положили конец диагностической одиссее для многих пациентов, страдающих хромосомными и моногенными заболеваниями. Эти методы расширили наши представления об уже известных заболеваниях и привели к открытию множества новых взаимосвязей между изменениями в геноме и патологическими состояниями.
ВОЗМОЖНОСТИ И ВЫЗОВЫ
Геном человека подобен тексту книги. Бесчисленные значки на поверхности страниц не имеют смысла, пока не интерпретированы читателем. Он должен обладать визуальным восприятием, владеть письменностью на данном языке и уметь преобразовывать абстрактные символы в мысленные образы с определенным содержанием и контекстом. Точно так же последовательность из нескольких миллиардов нуклеотидов в геноме человека бессмысленна, пока клеточные механизмы не интерпретируют ее в инструкции для синтеза белков или функциональных РНК. Эти инструкции невидимы и непонятны ученым, пока не будут обнаружены, переведены и интерпретированы сложными инструментами секвенирования и мощными компьютерными алгоритмами. Но, в отличие от книги, смысл которой человеку с интеллектом постичь нетрудно, когда дело доходит до чтения генома, мы до сих пор выглядим немногим лучше двухлетнего ребенка, пытающегося понять Британскую энциклопедию. Тем не менее шаги к постижению значения изменений в геноме уже сделаны. Революционными событиями последних десятилетий стали:
- завершение в 2003 году проекта «Геном человека», обеспечившего инструменты и справочную информацию для генетических исследований;
- появление хромосомного микроматричного анализа (ХМА) в начале и высокопроизводительного секвенирования (NGS) — в середине 2000-х годов, что позволило генетикам анализировать число копий ДНК и ее последовательность при приемлемой стоимости и времени обработки (рис. 1).
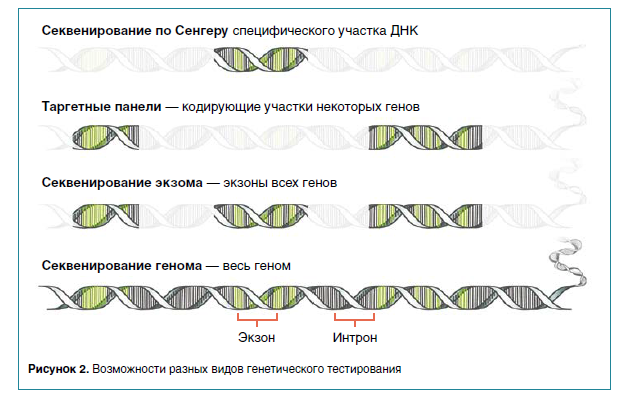
Однако этого недостаточно для клинического использования данных технологий. Лишь стремительный рост возможностей компьютерной обработки информации позволил применить ХМА и NGS для диагностики наследственных заболеваний. Компьютерные платформы для анализа геномных данных продемонстрировали впечатляющий диагностический результат. В последние годы для клинических целей применяется секвенирование генома, превосходящее по полноте охвата все ранее существовавшие методы генетического тестирования. В отличие от секвенирования экзома (совокупность экзонов всех генов), оно позволяет определить последовательность генов, включая интроны, а также регуляторных и межгенных участков генома (рис. 2).
Например, работа 14,5 тысячи генов, связанных с формированием и функционированием сердца, регулируется 55 тысячами промоторов и энхансеров. Оказалось, что именно в этих регуляторных элементах, а не в кодирующих частях генов, локализованы многие патогенные генетические варианты. Благодаря открывшимся возможностям лаборатории клинической геномики по всему миру начали сообщать о 25–50 % диагностической эффективности секвенирования генома. Это существенно выше, чем у любого другого генетического теста — молекулярного, цитогенетического или биохимического. Однако в ряде ситуаций ХМА, секвенирование экзома или генома не дают результатов или даже создают новые проблемы для пациентов и врачей, заставляя их менять клиническое мышление.
КОМПЛЕКСНЫЙ ДИАГНОЗ
Еще 20–30 лет назад считалось, что у человека не может быть более одного генетического заболевания. Умножив частоту одной редкой наследственной болезни на частоту другой, получим крайне малую величину, означающую ничтожно низкую вероятность сочетания двух таких патологий. Но в эпоху высокоразрешающих технологий двойной диагноз уже не редкость. У 4–15 % больных онаруживаются мутации в более чем одном гене, вызывающем заболевание. При этом каждый вариант дает частичное объяснение комплексной клинической картины пациента.
В практике даже одного врача-генетика возможны неоднократные случаи не только двойного, но и тройного диагноза. Мы наблюдали девочку с комплексным фенотипом — лицевыми признаками синдрома Корнелии де Ланге (длинные густые ресницы и брови, синофриз, длинный фильтрум), умственной отсталостью, судорогами и двигательными нарушениями, типичными для синдрома Ангельмана (атаксия, движения «механической куклы»), и нарушениями ритма сердца. При молекулярном кариотипировании получены доказательства синдрома Ангельмана — участок потери гетерозиготности, охватывавший гены PWRN2, PWRN1, C15orf2 (NPAP1), SNRPN, PWAR5 (arr 15q11.2 [24, 251, 567–25, 253, 314] х 2hmz).
При секвенировании экзома определены гетерозиготные патогенные варианты в еще двух генах: c. 3518 G>A (p.Cys1173Tyr) в гене KMT2A, связанном с синдромом Корнелии де Ланге, тип 1 (OMIM 122470) и c.1176 C>T (p.Arg3906Trp) в гене ANK2, связанном с синдромом удлиненного интервала QT тип 4 (OMIM 600919). Так сложный фенотип из признаков трех заболеваний нашел объяснение только после комплексного молекулярногенетического и молекулярно-цитогенетического обследования.
ОСМЫСЛЕНИЕ РЕЗУЛЬТАТОВ
Опытный врач-генетик бывает часто разочарован лабораторными отчетами, не подтверждающими диагноз, установленный им по клиническим признакам. У нас была пациентка, с раннего возраста наблюдавшаяся с неопределенным диагнозом. Разными генетиками предполагались синдромы Нунан и Костелло, поскольку девочка имела характерные признаки этих заболеваний. Для первого синдрома (тип 3) типичны задержка роста и стеноз легочной артерии, а для второго — макроцефалия, грубые черты лица, редкие тонкие волосы, глубокие ладонные борозды. Наблюдались и общие для этих заболеваний признаки: задержка речевого развития, широкий лоб, гипертелоризм, низкопосаженные ушные раковины, что затрудняло дифференциальную диагностику
Гены обоих синдромов принадлежат к одному и тому же молекулярному процессу — сигнальному пути RAS-MAPK. Он осуществляет передачу сигнала от внешней клеточной мембраны к ядру клетки в ответ на действие факторов роста. Вероятно, участие продуктов генов синдрома Костелло (HRAS) и синдрома Нунан, тип 3 (KRAS), в одном процессе и лежит в основе некоторого клинического сходства этих болезней.
Ребенку неоднократно проводили секвенирование по Сенгеру генов HRAS и KRAS— без результата. Генетики считали, что мутация в одном из этих генов есть, но находится в труднодоступном для секвенирования участке или регуляторной области, к которой не подобраться. Однако после секвенирования генома был обнаружен гетерозиготный генетический вариант, но не там, где его искали, а в гене SHOC2 (c.4A>G$ p.Ser2Gly). Ген SHOC2 ассоциирован с нунан-подобным синдромом с потерей волос (OMIM 607721), включающим все клинические признаки, наблюдавшиеся у девочки. А ген SHOC2 входит в ту же генную сеть, что и KRAS и HRAS, с которыми он функционально взаимосвязан. Все три гена относятся к сигнальному пути RAS-MAPK, а соответствующие им заболевания—к расопатиям.
Из таких случаев можно извлечь два урока. Во-первых, необходимо подавлять в себе высокомерие опытных специалистов и быть готовыми к тому, что предполагаемый нами клинический диагноз не подтвердится молекулярно. Второе и, возможно, более важное: со временем мы будем связывать определенные фенотипы не с каким-то одним геном, а с вовлечением определенных молекулярных процессов. В упомянутом случае нунанподобный синдром с потерей волос имел выраженное фенотипическое сходство с другими расопатиями. Их гены вовлечены в один и тот же сигнальный путь RAS-MAPK, что и затрудняло клиническую диагностику
НОЧНОЙ КОШМАР ГЕНЕТИКА
Еще до появления высокопроизводительного секвенирования, с внедрением секвенирования в клиническую практику возник термин «варианты неопределенного значения» (variants of uncertain significance, VUS). Как же решать проблему интерпретации десятков тысяч вариантов, не соответствующих эталонной последовательности генома, выявленных при секвенировании экзома, и миллионов вариантов, выявленных при секвенировании генома? Для этого члены Американской коллегии медицинской генетики (American College of Medical Genetics, ACMG) подготовили рекомендации по интерпретации новых вариантов последовательностей ДНК, не задокументированных ранее как вызывающие заболевание. Была разработана их классификация по 5-уровневой шкале: патогенный вариант, вероятно патогенный, неопределенного значения, вероятно доброкачественный, доброкачественный. Можно спорить об относительном значении таких факторов, как совпадение с базами данных, эволюционная консервативность затронутого аминокислотного остатка в белковом продукте гена, класс изменения последовательности ДНК (миссенсвариант, вариант потери функции, изменение сайта сплайсинга), степень фенотипического совпадения с проявлениями у пациента и т.д., но в целом классификация оказалась весьма полезной и получила широкое распространение.
Однако в ней есть неустраняемый элемент субъективности, что подтверждается противоречивыми интерпретациями одних и тех же вариантов разными экспертными лабораториями, использующими данные рекомендации. Это говорит о необходимости совместной работы опытных биоинформатиков и клиницистов. Вряд ли в ближайшее время их заменит полностью автоматизированный алгоритм или система искусственного интеллекта. Ведь специалисты хорошо понимают, что каждый пациент индивидуален и любые правила могут и должны нарушаться.
Термин VUS стал источником особого раздражения и для лабораторий, и для клиницистов, получающих лабораторные отчеты. VUS вполне может оказаться и патогенным, и доброкачественным: на момент сообщения просто недостаточно знаний, чтобы провести различие. Есть потенциальная опасность «переоценки» VUS, поскольку они могут использоваться для пренатальной диагностики при последующих беременностях, приводя к их необоснованному прерыванию или иным образом подталкивая врача в неверном направлении. С другой стороны, многие клиницисты недооценивают VUS, рассматривая их как «вероятно доброкачественные» изменения последовательности ДНК или, по крайней мере, «не требующие действий». Вспомним о негативном опыте США, когда при секвенировании по Сенгеру отдельно гена BRCA, связанного с раком груди, большинство пациентов, получивших результат класса VUS, предпочли не проводить профилактическую мастэктомию и даже не наблюдали регулярно за состоянием молочных желез, что привело к фатальным исходам во многих случаях. Для лечащего врача наличие VUS — стимул к дополнительному клиническому обследованию и наблюдению пациента в динамике. Необходимо сохранять информацию о VUS в медицинской карте пациента до появления в литературе новой информации о его клинических последствиях и наблюдать за тем, как разовьется фенотип пациента с возрастом. Биоинформатику следует осторожно использовать термин VUS в лабораторных отчетах и сопровождать его объяснением причин, позволяющими заподозрить возможную его связь с фенотипом пациента.
ВТОРИЧНЫЕ РЕЗУЛЬТАТЫ
Вторичные/случайные находки, подлежащие регистрации, наблюдаются примерно в 2 % случаев, то есть их количество огромно. В отличие от VUS, которые уже были знакомы лабораториям, выполняющим секвенирование отдельных генов или небольших панелей, «вторичные результаты»—это новое понятие, возникшее с появлением секвенирования всего экзома и генома. Вторичные находки в виде генетических вариантов, обнаруженных в определенных генах семейного рака или кардиомиопатий, могут оказаться летальными, но могут и не представлять непосредственной угрозы. Вторичные результаты по определению не имеют отношения к прямым показаниям для проведения NGS. Скорее, о них сообщают пациенту или его родителям для начала профилактических мероприятий. Это как случайно обнаружить опухоль легкого при рентгенографии грудной клетки, сделанной при подозрении на перелом ребра. Семья может быть полностью сосредоточена на актуальном для ребенка заболевании и не настроена воспринимать вторичный результат, указывающий на риск развития, например, рака груди через 40 лет. Можем ли мы принести больше вреда, чем пользы, усугубляя стрессовое состояние подобной семьи? Или как врачи мы обязаны «предупреждать», как и в случае обнаружения неожиданного симптома во время обычного медицинского осмотра?
Этот вопрос стал причиной долгих дебатов в сообществе медицинских генетиков, в результате вышло руководство ACMG по сообщению случайных/вторичных результатов. В нем перечислены 24 высокопенетрантных, потенциально летальных и тяжелых заболевания (семейный рак, кардиомиопатии, злокачественная гипертермия, туберозный склероз и др.), вызванных патогенными вариантами в 56 генах, которые необходимо искать и сообщать о них в каждом случае. Однако некоторые коллеги выступили против императивности руководства, не оставляющего ребенку или родителям возможности отказаться от информации о вторичных результатах, что превращает диагностический тест секвенирования в скрининговый тест на заболевания, возникающие в старшем возрасте. Большинство вторичных результатов из списка ACMG представляют собой аллели риска неопределенной пенетрантности. В этом их отличие от случайных находок на рентгенограмме грудной клетки, фиксирующих признаки текущей болезни. На - пример, пенетрантность «патогенных» вариантов внезапной сердечной смерти при синдромах удлиненного интервала QT, вероятно, не превышает 50 %. Надо ли обременять всех пациентов, у которых обнаруживается вторичная находка в этих генах, пугающей перспективой, учитывая, что они не проводили тест секвенирования по поводу нарушений ритма сердца и имеют нормальную ЭКГ? Все зависит от желания человека. Сообщение о вторичных находках осуществляется с информированного согласия пациента или его представителя. Если больной не хочет знать о предрасположенности к заболеванию — никто не вправе навязывать ему эти знания.
ГЕНЕТИЧЕСКИЙ СКРИНИНГ
Еще одна этическая проблема—неравенство доступа к тестированию. Если так важно сообщать пациенту о случайных находках при геномном секвенировании по поводу другого заболевания, то почему этот тест не предлагается в качестве скрининга риска для всей популяции? Должен ли человек сначала обнаружить у себя симптомы генетической патоогии, чтобы иметь право на скрининг геномного риска? Ответа пока нет, поскольку нет и установленных руководящих принципов генетического скрининга на перечисленные в списке AСMG заболевания среди населения в целом (детей и взрослых).
Между тем ведутся пилотные исследования геномного скрининга новорожденных. По мере снижения затрат на секвенирование некоторые формы скрининга новорожденных с помощью NGS станут стандартом в следующие 5–10 лет, хотя это вряд ли заменит биохимический скрининг тяжелых болезней обмена. Фактически мы уже применяем скрининговый тест с использованием NGS — это неинвазивный пренатальный скрининг для выявления анеуплоидий у плода. Эта новая технология как дополнение к биохимическому скринингу материнской сыворотки и альтернатива инвазивным амниоцентезу или биопсии ворсин хориона изменила мир пренатальной диагностики. Тест нравится и акушерам, потому что он информативнее скрининга материнской сыворотки, и пациентам, потому что его можно проводить на более ранних сроках беременности и без риска для плода по сравнению с амниоцентезом. Но не будем забывать, что это лишь скрининговый тест: его результат надо подтверждать инвазивными методами.
Другой тип генетического скрининга с использованием NGS также нашел применение в дородовых клиниках некоторых стран. Речь идет о расширенном скрининге носителей, который значительно распространился за пределы горстки генов и болезней, ранее предлагавшихся для скрининга парам определенного этнического происхождения (афроамериканцы, средиземноморцы, евреи-ашкенази и т.д.). Генетические исследования молодых супружеских пар, планирующих рождение детей, для оценки риска развития моногенных и хромосомных заболеваний и их профилактики с последующим применением медико-генетического консультирования, преимплантационной и пренатальной диагностики целесообразны экономически. Тревожит лишь будущая ответственность врачей-генетиков, которых попросят дать рекомендации для репродуктивных решений, порой необратимых, основываясь на рисках, о которых мы мало знаем, на базе крайне редких вариантов, которые недостаточно хорошо охарактеризованы, при расстройствах, естественное течение которых мало изучено, в том числе протекающих бессимптомно.
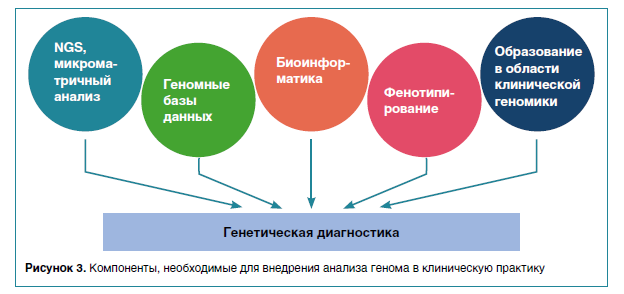
В контексте вторичных находок и неинвазивного пренатального скрининга, необходимо избегать объединения скрининговых тестов с диагностическими. Скрининг носителей, несмотря на то что в названии есть слово «скрининг», в конечном итоге приведет к диагностическому тесту у плода, основанному на результатах секвенирования.В целом внедрение высокоразрешающих методов дало толчок к установлению диагноза для пациентов с крайне редкими или до сих пор неизвестными наследственными заболеваниями (рис. 3). Даже если какое-то генетическое заболевание не имеет способа лечения (а именно так обычно и бывает), выявленный патогенный вариант можно использовать при медико-генетическом консультировании семьи и пренатальной диагностике во время будущих беременностей.
НАШИ ПАРТНЕРЫ