



Статьи
Синдром Альпорта: история длиною в век
В последние 10 лет к синдрому Альпорта (СА), проявляющемуся прогрессирующей нефропатией, нейросенсорной тугоухостью и патологией глаз, приковано пристальное внимание как специалистов, так и пациентских организаций. Огромные усилия направлены на поиски методов лечения, способных кардинально изменить прогноз заболевания таких больных. О достижениях в этой области и пойдет речь в статье.
ИСТОРИЯ ЗАБОЛЕВАНИЯ
Семейный случай геморрагического нефрита с прогрессирующим снижением функции почек впервые описан в 1923 г. Четырьмя годами позже южноафриканский врач Aртур Сесил Альпорт (1880– 1959) обратил внимание на сочетание нейросенсорной тугоухости и нефропатии у ранее описанных британских пациентов. Альпорт объединил два симптома (тугоухость и геморрагический нефрит) в синдром с предполагаемым аутосомно-доминантным характером наследования, названный впоследствии в его честь.
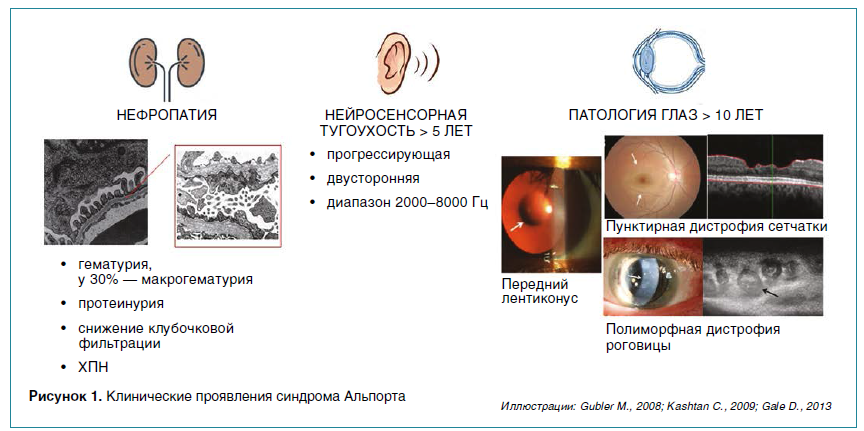
СА — наследственное прогрессирующее заболевание, вызванное мутацией в генах α3-, α4-, α5-цепей коллагена IV типа (COL4A3, COL4A4, COL4A5), характеризующееся развитием нефропатии в вариабельном сочетании с нейросенсорной тугоухостью и патологией органов зрения (рис. 1). Гены α3-, α4-цепей коллагена IV типа (COL4A3, COL4A4) локализованы на второй хромосоме. Гетеро- или гомозиготная мутация в них вызывает аутосомно-доминантный или аутосомно-рецессивный вариант СА соответственно. Патогенные варианты в гене, кодирующем α5‑цепь (COL4A5), расположенном на Х-хромосоме, ведут к развитию Х-сцепленного варианта заболевания. Как правило, оно передается из поколения в поколение. Лишь 10–15 % случаев СА обусловлены спонтанными мутациями.
Исторически считается, что распространенность синдрома в среднем составляет один случай на 5000–10 000 человек. Однако эти данные не учитывают женщин с Х-сцепленным вариантом заболевания (которых до недавнего времени рассматривали как носителей мутаций в гене COL4A5), а также пациентов с нетипичной клинической картиной и изолированной доброкачественной микрогематурией. Согласно результатам секвенирования экзома 200 000 здоровых людей (Genomics England Research Consortium), опубликованным в 2021 г., распространенность патогенных вариантов в генах COL4A3 и COL4A4 в популяции составляет 1 случай на 200 человек, патогенных вариантов в гене COL4A5 – 1 на 2000 человек. Это значит, что частота СА значительно выше предполагавшейся ранее. И если в России, согласно данным Росстата за 2021 г., проживает около 145 млн человек, включая 33 млн детей и подростков (0–19 лет), то более миллиона жителей нашей страны, в том числе около 350 тыс детей и подростков имеют СА.
В структуре наследственной нефрологической патологии у детей СА занимает второе место после аутосомно-доминантной поликистозной болезни почек, отвечая за 0,3–2,2 % всех случаев терминальной хронической почечной недостаточности в молодом возрасте.
Развитие СА связано с нарушением синтеза α3-, α4-, α5-цепей коллагена IV типа, которые образуют гетеротримеры, составляющие основу базальной мембраны почечных клубочков, хрусталика, сетчатки и роговицы глаза, улитки внутреннего уха и легких. Снижение или отсутствие синтеза одной из α-цепей приводит к изменению структуры базальных мембран, их химической и механической нестабильности, нарушению их функции с течением времени.
ОРГАН-МИШЕНЬ
Основным проявлением СА, определяющим прогноз пациентов, служит заболевание почек. Гетеротримеры α3-, α4-, α5‑цепей коллагена IV типа составляют основу базальной мембраны почечных клубочков, осуществляющих фильтрацию. Ранний симптом нефропатии — постоянная гломерулярная гематурия (более 80 % дисморфных эритроцитов в осадке мочи) разной степени выраженности. Это объясняется способностью эритроцитов проникать через дефекты в базальной мембране клубочков. Современные исследования показывают, что причиной гломерулярной микрогематурии у детей, в том числе при отсутствии семейного анамнеза, является прежде всего СА: по данным детской клиники Техаса, на его долю приходится около половины случаев гематурии. У некоторых пациентов на фоне интеркуррентных заболеваний возникают эпизоды макрогематурии. Согласно данным нашей клиники, макрогематурия встречается у трети детей с Х-сцепленным СА, преимущественно в возрасте от 1 до 4 лет, с одинаковой частой у мальчиков и девочек. По мере прогрессирования заболевания почек к гематурии последовательно присоединяются альбуминурия, протеинурия и снижение клубочковой фильтрации.
Прогрессирование нефропатии при СА связывают с растяжением «дефектных» гломерулярных базальных мембран, не справляющихся с нагрузкой нормальным внутрикапиллярным давлением, с активацией всех клеток почечных клубочков (эндотелиальных, мезангиальных, подоцитарных), на которые передается это избыточное давление, с синтезом цитокинов и белков, направленных на восстановление поврежденной базальной мембраны.
Механизм повреждения гломерул легче понять, если представить гломерулярный капилляр в виде шланга, внутренний слой которого выстлан эндотелием, средний представлен армированной сеткой, состоящей из гетеротримеров коллагена, а наружный образован подоцитами. При подаче жидкости в шланг с дефектным армированным каркасом растягивается и внутренний слой, лишенный опоры, и наружный, подвергающийся непривычному давлению. В начальной стадии компенсаторные механизмы способны поддерживать нормальную функцию капилляра клубочка. Но ни один из множества синтезируемых матричных белков (ламинин, α1- и α2‑цепи коллагена IV типа, нидоген и др.) не может сравниться по прочности с α3-, α4-, α5‑гетеротримерами. Поэтому, когда фильтрационная нагрузка на гломерулы физиологически увеличивается с возрастом, компенсаторных механизмов становится недостаточно. При микроскопии капилляры клубочков выглядят растянутыми, с утолщенными рыхлыми и дезорганизованными базальными мембранами. Подоциты, пытаясь покрыть все более увеличивающуюся площадь «растянутых» капилляров, гипертрофируются, их ножки распластываются, утрачивают связь с базальной мембраной.
В результате потери подоцитов и избыточного синтеза матричных белков развивается склероз гломерул и интерстиция, сопровождающийся появлением протеинурии и снижением фильтрационной функции почек.

Выделяют несколько стадий нефропатии при СА, имеющих клинические и морфологические особенности (табл. 1):
- изолированная гематурия, которой на морфологическом уровне соответствуют тонкие гломерулярные базальные мембраны, сохранные подоциты;
- стадии (1) альбуминурии и (2) протеинурии, когда отмечаются очаговое утолщение/расслоение базальных мембран клубочков и подоцитопатия;
- стадии (3 и 4) снижения фильтрационной функции почек с диффузным утолщением и расслоением гломерулярных базальных мембран и гломерулосклерозом разной степени выраженности.
Исходя из патогенеза, очевидно, что на темпы прогрессирования нефропатии при СА влияют:
- степень нарушения синтеза α3-, α4-, α5‑цепей коллагена (характер мутации генов COL4A3, COL4A4, COL4A5);
- функциональные особенности подоцитов и их способность к адаптации (наличие других генетических вариантов/ полиморфизма подоцитарных генов);
- факторы, способствующие повышенной нагрузке на гломерулы или гиперфильтрации (относительное/абсолютное уменьшение количества гломерул: недоношенность тяжелой степени, ожирение, единственная почка и другие);
- сопутствующие острые и хронические заболевания почек, приводящие к повреждению гломерул за счет других механизмов (гломерулонефрит, диабетическая нефропатия);
- артериальная гипертензия (АГ) и другие факторы кардиоваскулярного риска.
Для улучшения нефрологического прогноза пациентов необходимо устранить все модифицируемые факторы риска прогрессирования нефропатии и как можно раньше снизить внутриклубочковое давление.
СЛУХ И ЗРЕНИЕ
Наряду с нефропатией 30–50 % пациентов с СА имеют двустороннюю нейросенсорную тугоухость, которая обычно развивается в раннем школьном возрасте у мальчиков с Х-сцепленным и у всех пациентов с аутосомно-рецессивным вариантами заболевания, а у женщин с Х-сцепленным и пациентов с аутосомно-доминантным вариантами — после 35 и 60 лет соответственно. Потеря слуха затрагивает прежде всего высокочастотный звуковой диапазон (дети плохо слышат пение птиц, звон колокольчика, женские голоса) и прогрессирует с возрастом.
Предполагается, что тугоухость обусловливают:
- снижение натяжения базилярной мембраны улитки уха из-за слабости спиральной связки;
- растяжение дефектной базальной мембраны сосудистого слоя внутреннего уха с нарушением ее функции.
Развитие патологии глаз также связано с уменьшением механической резистентности мембран капсулы хрусталика (передний/задний лентиконус), сосудистой оболочки глаз (ретинопатия), боуменовой и десцеметовой мембран роговицы (ее дистрофия и эрозия).
Офтальмологические симптомы — менее чувствительный (так как встречается редко), но более специфичный (более надежный для постановки диагноза) признак заболевания по сравнению с нейросенсорной тугоухостью. Выявление типичной для СА патологии органов зрения характерно для более тяжелых вариантов болезни: Х-сцепленного у мальчиков и аутосомно-рецессивного у всех пациентов независимо от пола.
Формирование лентиконуса может проявляться нарушением фокуса зрения и в некоторых случаях осложняться спонтанным разрывом капсулы хрусталика с формированием катаракты. Периферическая пунктирная ретинопатия выявляется у большинства мужчин и у 25 % женщин с Х-сцепленным СА, она также характерна для пациентов с аутосомноцессивным вариантом заболевания. Эрозии роговицы обнаруживаются примерно у 10 % пациентов в позднем подростковом возрасте, иногда их появление предшествует диагностике СА. Задняя полиморфная дистрофия роговицы, связанная с истончением десцеметовой мембраны субэндотелия, может сопровождаться светобоязнью, слезотечением, ощущением инородного тела в глазу. Заболевание диагностируется при исследовании передней камеры глаза методами оптической когерентной томографии высокого разрешения, конфокальной микроскопии, биомикроскопии в щелевой лампе.
У пациентов с СА могут быть аномалии крупных сосудов:
- дилатация аорты, аневризмы ее нисходящего отдела, внутримозговых и коронарных артерий;
- пролапс митрального и недостаточность аортального клапанов,
- дефекты межжелудочковой перегородки.
К крайне редким клиническим проявлениям у пациентов с СА относятся симптомы, связанные с продленными делециями генов COL4A5-COL4A6 и COL4A5-FACL4-AMMECR1. В первом случае у больных развивается лейомиоматоз (доброкачественные гладкомышечные опухоли) желудочно-кишечного тракта (прогрессирующая дисфагия, боли за грудиной и рвота после еды, диспепсия), патология бронхолегочной (рецидивирующие бронхиты, кашель, одышка) и женской репродуктивной систем. Во втором случае у пациентов отмечаются гипоплазия лица с недоразвитием глазниц, скул и верхней челюсти, дефицит интеллекта и эллиптоцитоз (овальные эритроциты).
ВАРИАНТЫ НАСЛЕДОВАНИЯ
Известны три основных варианта наследования СА (рис. 2):
- Х-сцепленный, выявляемый у 80–85 % пациентов и обусловленный мутациями в гене COL4A5;
- аутосомно-рецессивный, связанный с вариантами в генах COL4A3 или COL4A4;
- аутосомно-доминантный, на долю которого приходится около 10–15 % пациентов с СА.
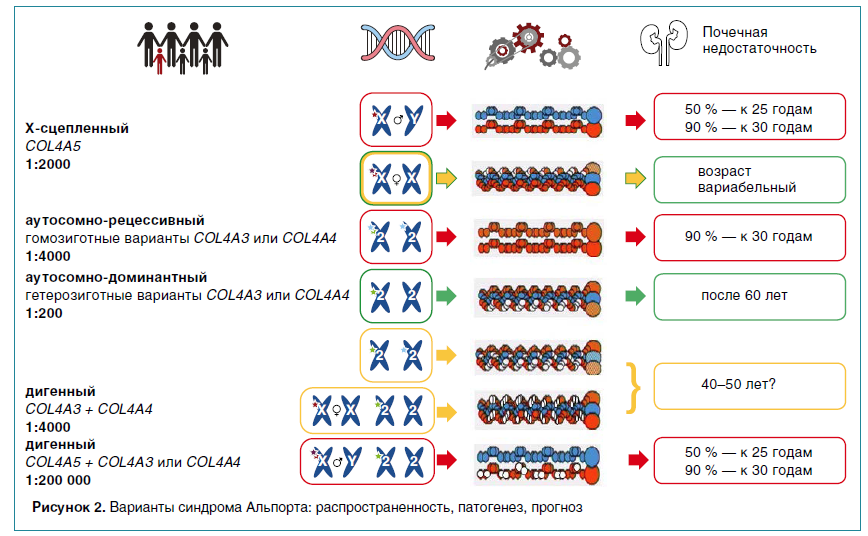
В 2015 г. международная группа авторов опубликовала данные о дигенном наследовании заболевания, выявляемом приблизительно у 1 % пациентов с СА и обусловленном сочетанными мутациями в разных генах коллагена IV типа: COL4A3 и COL4A4, COL4A5 и COL4A3 или COL4A4.
Х-сцепленный вариант заболевания характеризуется более тяжелой клинической картиной у мужчин, с прогрессирующим течением нефропатии, неизбежно приводящей к развитию терминальной почечной недостаточности во второй-третьей декадах жизни и нейросенсорной тугоухости более чем в половине случаев. У женщин чаще встречается изолированный мочевой синдром (20 % — изолированная гематурия, 75 % — гематурия с протеинурией), почечная недостаточность и снижение слуха развиваются менее чем в половине случаев (42 %), как правило, после 55 лет. Учитывая наличие клинических проявлений заболевания у женщин, сейчас не используется термин «носительство» при выявлении у них гетерозиготной мутации в COL4A5.
Течение нефропатии у женщин с Х-сцепленным СА сложно предсказать из-за спонтанной инактивации одной из Х-хромосом и связанного с этим мозаичного синтеза α5‑цепи в разных тканях и даже в разных клетках в пределах одного органа (например, в подоцитах в пределах одной гломерулы). Тяжесть клинических проявлений будет зависеть от степени инактивации Х-хромосомы, несущей мутацию. Потому все пациентки с Х-сцепленным СА с детства нуждаются в динамическом наблюдении, нефропротективной терапии и в большинстве случаев не могут быть потенциальными родственными донорами почки.
Клинические проявления и прогноз пациентов с аутосомно-рецессивным вариантом СА не зависят от пола и соответствуют таковым у мужчин с Х-сцепленным типом наследования болезни. Аутосомно-доминантная форма СА характеризуется наиболее благоприятным течением: как правило, экстраренальные проявления отсутствуют, терминальная стадия почечной недостаточности развивается лишь у 20–30 % пациентов после 60 лет.
J. Savige с коллегами показала, что при дигенном наследовании наличие гетерозиготных мутаций в COL4A3 или COL4A4 у пациентов с Х-сцепленным СА ухудшает прогноз заболевания у девочек и практически не влияет на нефрологический прогноз у мальчиков. У пациентов с сочетанием гетерозиготных вариантов в COL4A3 и COL4A4 более неблагоприятный прогноз по сравнению с аутосомно-доминантным и более благоприятный по сравнению с аутосомно-рецессивным типом СА.
При развитии терминальной почечной недостаточности пациентам проводят трансплантацию почки, при этом выживаемость почечного транслантата при СА лучше, чем при других заболеваниях почек, частота нефрита, ассоциированного с образованием антител к гломерулярной базальной мембране, не превышает 5 %.
Описано около 3000 вариантов мутаций в генах COL4A3, COL4A4 и COL4A5, абсолютное большинство которых уникальны. Только 12 вариантов в гене COL4A5 были зафиксированы в неродственных семьях более 5 раз с преобладанием в отдельных географических регионах мира. Например, вариант COL4A5 p.Gly624Asp, вызывающий замену глицина на аспарагин в коллагеновом домене гена, является самым распространенным в Европе, включая Россию (более 30 % семей с миссенс-вариантами), и обуславливает относительно благоприятное течение нефропатии с более поздним развитием почечной недостаточности по сравнению с другими миссенс-вариантами: 54 [50; 62] и 26 [22; 30] лет соответственно.
У мужчин с Х-сцепленным вариантом болезни установлена четкая зависимость фенотипических проявлений заболевания от типа и локализации мутации в гене COL4A5. Генетические варианты, программирующие преждевременную терминацию синтеза белка (большие перестройки, нонсенс-мутации и сдвиг рамки считывания), ассоциируются с развитием почечной недостаточности во второй декаде жизни. При миссенс-вариантах почечная недостаточность, как правило, развивается примерно в 40 лет. Пациенты с вариантами, затрагивающими сайты сплайсинга, имеют промежуточную почечную выживаемость. Нефрологический прогноз хуже при мутациях в области, кодирующей сигнальный пептид, по сравнению с коллагенновым и неколлагеновым NC1‑доменами гена COL4A5. У женщин зависимость клинических проявлений СА от характера мутации в COL4A5 менее очевидна.
В некоторых случаях у пациентов с СА отмечается несоответствие тяжести проявлений заболевания характеру выявленной мутации COL4A5. Так, например, делеции в гене COL4A5, кратные триплету оснований, приводящие вместо полного отсутствия синтеза к выработке белка с измененными свойствами, соматический мозаицизм по Х-хромосоме улучшают прогноз.
Раннее появление и высокий уровень протеинурии, быстрое снижение функции почек, раннее развитие тугоухости могут быть обусловлены миссенс-мутациями, при которых синтезируемый белок неспособен транспортироваться из клетки, а также сочетанием мутаций в генах α-цепей коллагена IV типа и подоцитарных генах, наличием гомозиготной мутации COL4A5 у женщин.
ВОПРОСЫ ДИАГНОСТИКИ
С 1974 г. разные группы исследователей предлагали критерии диагностики СА. Наиболее широкое распространение получили критерии, разработанные F.A. Flinter и соавторами. Для диагностики заболевания необходимо, чтобы у пациента с гематурией были три из четырех следующих признаков:
- Семейный анамнез, отягощенный по микро-/макрогематурии и/или почечной недостаточности;
- Типичные изменения базальных мембран клубочков при электронной микроскопии;
- Типичная патология глаз (передний лентиконус, макулярная дистрофия);
- Высокотональная нейросенсорная тугоухость.
Однако сопоставление результатов обследования пациентов с генетически подтвержденным СА показало, что клинически диагноз может быть поставлен только одной трети пациентов.
В 2013 г. международная группа экспертов выделила:
- критерии, позволяющие заподозрить СА у пациента с постоянной гломерулярной гематурией:
- типичный семейный анамнез или случаи почечной недостаточности в семье при отсутствии других возможных причин ее развития или
- специфичные клинические симптомы у пробанда (нейросенсорная тугоухость, лентиконус, ретинопатия);
- критерии, которые подтверждают диагноз:
- расслоение гломерулярных базальных мембран или
- выявление патогенных мутаций в COL4A3, COL4A4, COL4A5.
Однако расслоение базальных мембран клубочков появляется по мере прогрессирования нефропатии (как правило, на стадии протеинурии), а прогноз у пациентов с СА определяется сроками назначения нефропротективной терапии независимо от характера мутации. Это значит, что генетическое исследование имеет первостепенное значение для ранней диагностики заболевания.
Генетический анализ позволяет:
- рано установить диагноз;
- назначить нефропротективную терапию;
- прогнозировать течение болезни и сроки развития почечной недостаточности;
- определить риск развития нефрита, обусловленного образованием антител к гломерулярной базальной мембране в почечном трансплантате;
- провести каскадное обследование членов семьи;
- осуществить семейное генетическое консультирование;
- определить возможность родственного донорства.
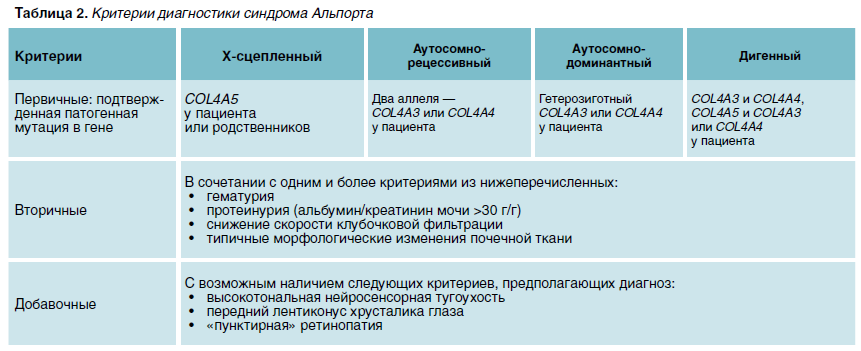
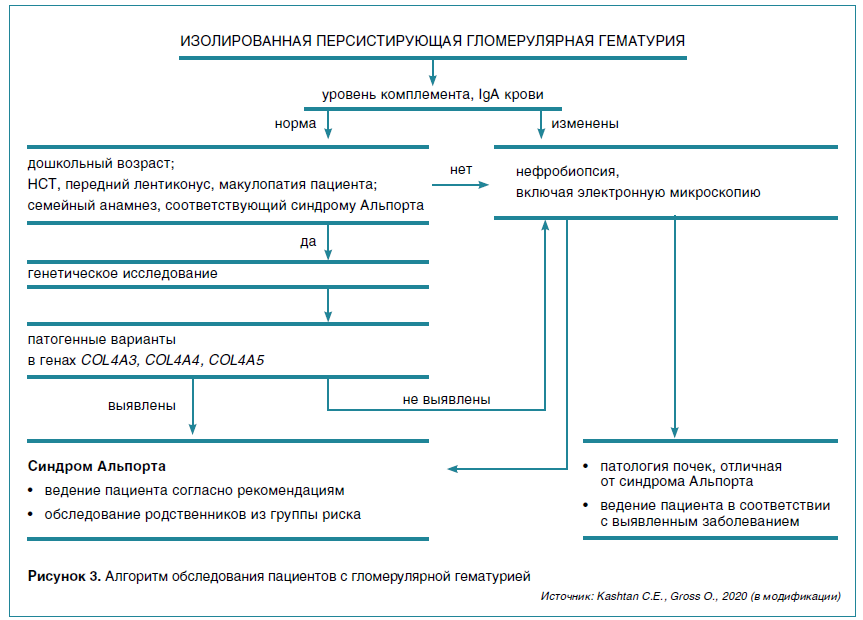
Вот почему сегодня пересмотрены критерии диагностики СА (табл. 2) и подходы к обследованию детей с изолированной гематурией — основным проявлением данного заболевания (рис. 3), согласно которым правильное обследование пациентов с изолированной гломерулярной гематурией и клинической картиной/семейным анамнезом, соответствующими СА, должно начинаться с генетического исследования.
ТЕРАПЕВТИЧЕСКИЕ ПОДХОДЫ
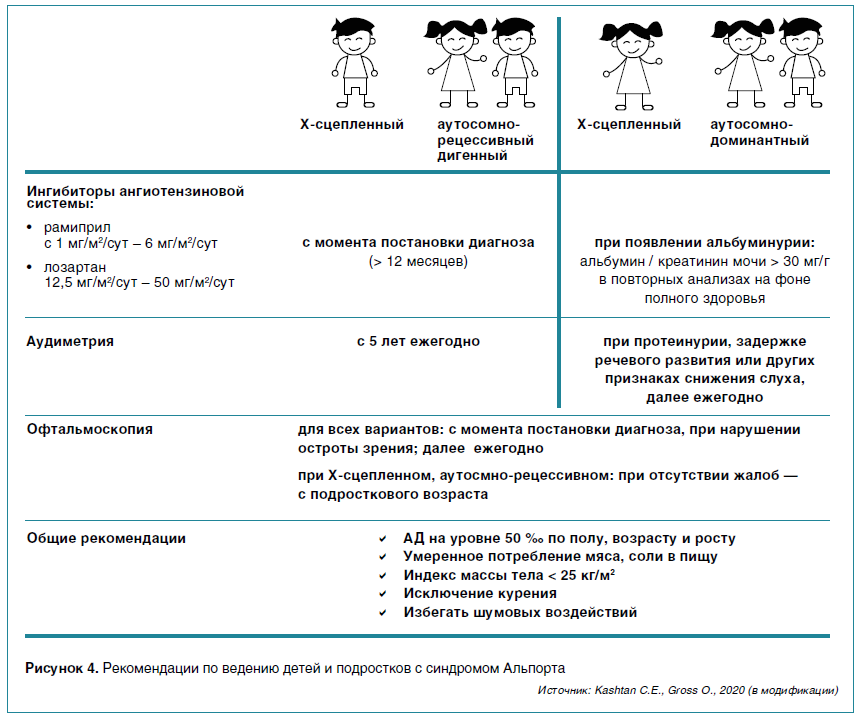
В 2011 г. немецкая группа во главе с O. Gross опубликовала данные о безопасности и эффективности использования ингибитора ангиотензин-превращающего фермента рамиприла в снижении темпов прогрессирования нефропатии при СА, которые легли в основу первых клинических рекомендаций по ведению пациентов (2013 г.). Сегодня больным из группы высокого риска развития почечной недостаточности в молодом возрасте при наличии альбуминурии, а также всем пациентам при наличии протеинурии (белок мочи > 4 мг/м2/час или > 0,2 мг/мг креатинина) назначаются off-label ингибиторов ангиотензин-превращающего фермента или блокаторы рецепторов к ангиотензину II типа. Плацебо-контролируемое исследование Early Protect по применению рамиприла (4,5–4,8 мг/м2/сут) на протяжении 6 лет у пациентов с СА, у которых была микрогематурия в сочетании с альбуминурией или без нее, показало снижение темпов прогрессирования нефропатии у 40 % больных на фоне лечения. Учитывая данные результаты, всем детям из группы риска рекомендуется назначать нефропротективную терапию с момента постановки диагноза (рис. 4). При ведении пациентов с СА важно обращать внимание на сопутствующие модифицируемые факторы риска прогрессирования патологии почек, особенно АГ. В современных рекомендациях предложена схема офтальмологического и аудиометрического обследования пациентов с учетом возрастных рисков появления экстраренальных симптомов.
Современная терапия позволяет замедлить темпы снижения функции почек при СА. Независимо от характера мутаций лечение ингибиторами ангиотензиновой системы увеличивает почечную выживаемость в среднем на 12 лет. Однако терапия блокаторами ангиотензиновой системы не влияет на прогрессирование нейросенсорной тугоухости. Экспериментально было показано, что ингибиторы эндотелина-А замедляют развитие тугоухости и уменьшают протеинурию, что делает их перспективными в терапии пациентов с СА. Сейчас исследуется эффективность ингибитора эндотелина-А — атрасентана (Affinity) и комбинированного препарата ингибиторов эндотелина-А и рецепторов ангиотензина — спарсентана (Duet и Duplex) при протеинурических вариантах нефропатий. Другим потенциальным методом лечения нефропатии является использование блокаторов глюкозо-натриевых котранспортеров II типа: исследование включило небольшое количество больных СА с фокально-сегментарным гломерулосклерозом и показало снижение протеинурии на 40 % от исходного уровня через 4,5 мес терапии.
Огромные усилия направлены на поиски методов лечения, способных кардинально изменить прогноз заболевания для пациентов с СА. По инициативе и благодаря поддержке пациентских организаций США, Англии, Франции, Италии были созданы научные консультативные группы и инициированы исследования по изучению данной патологии. С 2013 г. проводятся международные симпозиумы с участием клиницистов, ученых, представителей пациентов и фармацевтических компаний для обмена знаниями, определения направлений и объединения усилий в исследованиях.
Современные достижения в области генной терапии подарили надежду и пациентам с СА. Экспериментально продемонстрирована эффективность прямой (модифицирующая экспрессия генов COL4A3, COL4A4, COL4A5) и непрямой (модифицирующая экспрессия генов, влияющих на прогрессирование заболевания: LAMA2, COL4A6) генной терапии СА, основанной на CRISPR/Cas9‑активирующей и ингибирующей технологиях. Основной задачей на сегодняшний день является разработка методов доставки CRISPR/ Cas9‑комплекса непосредственно в почки.
Как и в других областях медицины, только объединение наших усилий позволит справиться с задачами, которые до недавнего времени казались нерешаемыми, и открыть новую эпоху для пациентов с СА.
Автор выражает признательность коллегам
и пациентам НИКИ педиатрии им. акад. Ю.Е. Вельтищева,
J. Savige, O. Gross, J. Boeckhaus, D. Gale, S. Daga,
S. Gear, A. Cooper и Alport Syndrome Alliance
за предоставленные материалы и поддержку.
НАШИ ПАРТНЕРЫ















