



Статьи
Болезнь Ниманна — Пика (тип А) у ребенка пяти месяцев
Болезнь Ниманна — Пика — редкое наследственное заболевание из группы лизосомных болезней накопления. Впервые описана педиатром Альбертом Ниманном в 1914 г. у ребенка с гепатоспленомегалией и прогрессирующим поражением центральной нервной системы (ЦНС). Впоследствии Людвиг Пик провел несколько патологоанатомических исследований пациентов с тяжелой неврологической симптоматикой и гепатоспленомегалией, погибших в раннем возрасте. Диагноз он обозначил как липоидноклеточную спленомегалию.
КЛИНИЧЕСКИЕ ФОРМЫ
Распространенность болезни Ниманна — Пика составляет от 0,4 до 0,6 случая на 100 тысяч новорожденных, наиболее часто она обнаруживается у евреевашкенази. Заболевание обусловлено патогенными вариантами гена SMPD1, которые приводят к недостаточности кислой сфингомиелиназы и накоплению сфингомиелина в лизосомах. Тип наследования — аутосомно-рецессивный.
В зависимости от возраста манифестации, тяжести течения и прогрессирующего поражения различают три клинические формы болезни Ниманна — Пика:
- тип А — инфантильная нейровисцеральная с манифестацией в первые месяцы жизни, быстрым прогрессирующим течением и ранним летальным исходом;
- тип А/В — хроническая нейровисцеральная с манифестацией на первом году жизни и более медленным прогрессированием неврологический симптоматики;
- тип В — хроническая висцеральная форма, манифестирующая в детском возрасте и протекающая без поражения ЦНС.
Основные клинические проявления болезни Ниманна — Пика (тип А):
- начало в первые месяцы жизни;
- задержка физического развития;
- у детей отмечается вялое сосание, трудности с кормлением, диарея, иногда рвота;
- иктеричность склер, кожи, иногда — ксантомы;
- большой выступающий живот, гепатоспленомегалия;
- частые респираторные инфекции;
- поражение легких, включая диффузные инфильтраты, аспирационные пневмонии;
- фиброз печени, портальная гипертензия, холестаз;
- симптом «вишневой косточки» при офтальмологическом обследовании;
- диффузная мышечная гипотония, мышечная слабость, прогрессирующая неврологическая симптоматика;
- лимфаденопатия.
При лабораторном исследовании выявляются микроцитарная анемия, гипербилирубинемия, повышение активности печеночных трансаминаз, дислипидемия. В биоптатах костного мозга — крупные вакуолизированные пенистые клетки (NP cells), гистиоциты цвета морской волны.
У пациентов с болезнью Ниманна—Пика при проведении лабораторной диагностики отмечается резкое снижение или отсутствие активности сфингомиелиназы в высушенных пятнах крови, лейкоцитах и культуре кожных фибробластов. Для подтверждения диагноза показано молекулярно-генетическое обследование с целью выявления патогенных вариантов в гене SMPD1.
ПРИМЕР ИЗ ПРАКТИКИ
Мальчик пяти месяцев поступил в клинику с жалобами на плохую прибавку массы тела, неустойчивый стул, увеличение живота, задержку моторного развития, вялость, нистагм слева. Семейный анамнез не отягощен, родители не родственники (позже аналогичное заболевание выявлено у младшей сестры пробанда).
Анамнез жизни. Ребенок от третьей беременности с угрозой прерывания на всем протяжении. Роды вторые, самостоятельные, в срок. Вес при рождении — 3050 г, длина тела — 52 см, оценка по шкале Aпгар — 8/9 баллов. Со вторых суток до конца первого месяца жизни отмечалась физиологическая желтуха, в связи с чем получал необходимую терапию. В 21 день проконсультирован неврологом, диагноз: «Синдром вегето-висцеральных дисфункций, мышечная дистония с тенденцией к гипертонусу, тремор подбородка и конечностей».
С двух месяцев жизни недостаточно прибавляет в весе, в связи с чем переведен на искусственное вскармливание. В 2,5 месяца повторно осмотрен неврологом, который отметил появление диффузной мышечной гипотонии. Тогда же перенес острый обструктивный бронхит, острый двусторонний сальпингоотит, а в три месяца — острую сегментарную правостороннюю внебольничную пневмонию.
У ребенка отмечалась задержка моторного развития, голову стал держать с четырех месяцев, неуверенно. В возрасте 4,5 месяца педиатр обратил внимание на увеличение размеров печени и селезенки. По данным ультразвукового исследования органов брюшной полости (УЗИ ОБП) выявлены эхо-признаки гепатомегалии, умеренное повышение эхогенности паренхимы печени, увеличение размеров селезенки, диффузные изменения поджелудочной железы. Направлен на госпитализацию в федеральный педиатрический центр.
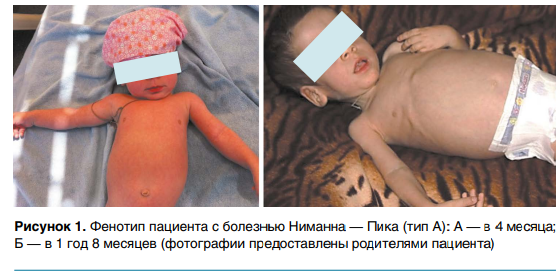
При осмотре в пятимесячном возрасте вес — 6480 г (10–25-й перцентили), длина тела — 66 см (75–90-й перцентили), окружность головы — 44,5 см (90–97-й перцентили), окружность груди — 39,5 см (210-й перцентиль), большой родничок— 2,5 × 2,5 см. Макроцефалия, грубоватые черты лица, высокий выступающий лоб, густые брови, длинные ресницы, широкая запавшая переносица, вздернутый нос, полные губы, низко расположенные ушные раковины (рис. 1). Ограничение подвижности коленных суставов. Диффузная мышечная гипотония, гипотрофия мышц конечностей. Голову держит неуверенно, не ползает, при вертикализации опора на ноги слабая. За предметами следит, отмечается ограничение движения глазных яблок кнаружи, вверх, вниз, периодически — горизонтальный мелкоразмашистый нистагм в крайних отведениях. На осмотр реагировал с интересом, улыбался, гулил, перекладывал и захватывал игрушки. В легких дыхание жесткое. Частота дыхательных движений (ЧДД) — 30 в минуту, частота сердечных сокращений (ЧСС)—125 ударов в минуту. Печень выступает на 3 см от края реберной дуги, селезенка—на 2 см.
Лабораторное обследование выявило значительное повышение активности печеночных трансаминаз, гаммаглютамилтранспептидазы, щелочной фосфатазы, лактатдегидрогеназы, концентрации холестерина и триглицеридов. Концентрации глюкозы, лактата, альфа-1-антитрипсина в крови были в норме. В общем анализе крови отмечался умеренно выраженный лейкоцитоз, анемия и тромбоцитопения не выявлены.
Инструментальные исследования
Нейросонография — признаки вентрикуломегалии, расширения субарахноидальных пространств и межполушарной щели.
УЗИ ОБП и почек — эхографические признаки гепатоспленомегалии, диффузного паренхиматозного процесса в печени, увеличение обеих почек.
Транзиентная эластография печени—умеренный фиброз (стадия F2 по шкале METAVIR)При проведении эхокардиографии патологических изменений не выявлено.
Рентгенография коленных суставов в прямой и боковой проекциях — рентгенологические признаки могут соответствовать минимальным проявлениям рахитических изменений.
При обследовании в стационаре диагностирована правосторонняя верхнедолевая (аспирационная) пневмония. Ребенок проконсультирован пульмонологом, назначена антибактериальная терапия.
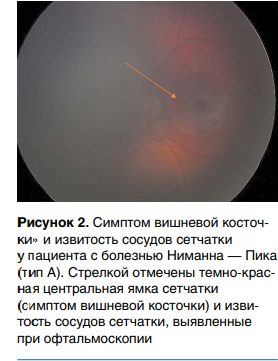
При офтальмологическом обследовании на глазном дне выявлен симптом вишневой косточки (рис. 2).
Проведена энзимодиагностика лизосомных болезней накопления. Обнаружено резкое снижение активности сфингомиелиназы в лейкоцитах крови — 0,02 нМ/мг/ч (норма — 0,56–3,24 нМ/мг/ч). Данные изменения характерны для болезни Ниманна — Пика (тип А).
Методом прямого автоматического секвенирования были исследованы все кодирующие экзоны и прилегающие к ним интронные области гена SMPD1 как у пациента, так и у его родителей.
По данным клинической картины и результатам молекулярно-генетического обследования установлен диагноз: «Болезнь Ниманна — Пика, тип А. Тип наследования—аутосомно-рецессивный».
В течение жизни у пациента прогрессировала неврологическая симптоматика: отмечались диффузная мышечная гипотония, мышечная слабость, формировались контрактуры в суставах, судорожный и псевдобульбарный синдромы. В 3 года в результате декомпенсации (по данным медицинской документации) легочносердечной недостаточности, возникшей как осложнение основного заболевания, зафиксирован летальный исход.
Болезнь Ниманна — Пика (тип А) — редкая патология из группы лизосомных болезней накопления, манифестирующая в первые месяцы жизни плохой прибавкой массы тела, гепатоспленомегалией, поражением дыхательной системы, диффузной мышечной гипотонией, прогрессирующим поражением ЦНС.
Важный диагностический признак заболевания — обнаружение симптома вишневой косточки при офтальмологическом обследовании. Рецидивирующие пневмонии относятся к тяжелым, прогностически неблагоприятным клиническим проявлениям заболевания, что в сочетании с прогрессирующим поражением ЦНС и гепатобилиарной системы приводит к летальному исходу.
Дифференциальная диагностика болезни Ниманна — Пика (тип А) проводится с болезнями Гоше и Вольмана (дефицит лизосомной кислой липазы), инфантильными формами GM1- и GM2-ганглиозидозов, муколипидозом (тип II), тяжелыми формами мукополисахаридозов (МПС I, МПС VII), галактосиалидозом, тирозинемией (тип 1), недостаточностью альфа-1-антитрипсина, муковисцидозом, болезнью Вильсона—Коновалова, гликогеновой болезнью IV типа (болезнь Помпе, инфантильная форма), врожденными нарушениями гликозилирования (тип IIL).
Лечение болезни Ниманна — Пика (тип А) включает в себя симптоматическую терапию, направленную на стабилизацию состояния пациента. В настоящее время разработаны методы патогенетической терапии всех трех типов болезни Ниманна—Пика (А, А/В и В), применение которых, однако, при тяжелом течении заболевания ограничено. Ферментозаместительная терапия рекомбинантной кислой сфингомиелиназой (олипудаза альфа) используется при лечении пациентов с болезнью Ниманна — Пика (тип В). Имеются описания пациентов с болезнью Ниманна—Пика (тип А/В), у которых на фоне ферментозаместительной терапии отмечалась положительная динамика. Перспективным методом лечения болезни Ниманна—Пика (тип А) является генная терапия.
Список литературы находится в редакции
НАШИ ПАРТНЕРЫ















