Статьи
Роль генетики в детской онкологии
Своевременная диагностика и лечение злокачественных новообразований (ЗНО) детского возраста — одно из приоритетных направлений современной педиатрии. Нередко дети имеют наследственную предрасположенность к развитию ЗНО, что часто сопряжено с их ранней манифестацией и повышенным риском развития вторых опухолей. Поговорим о наиболее распространенных синдромах предрасположенности к ЗНО у педиатрических пациентов.
СИНДРОМАЛЬНЫЙ ПАЦИЕНТ
В практике педиатра могут встречаться пациенты с известными генетическими синдромами, имеющими отличительные фенотипические черты (синдромы Дауна и Ниймегена, пигментная ксеродерма и др.). В ряде случаев у таких больных отмечен повышенный риск развития ЗНО, в том числе в детском возрасте (табл. 1), что определяет необходимость пожизненного наблюдения с выполнением комплекса лабораторных и инструментальных методов исследования, а также консультации специалистов. При этом одни генетические синдромы обусловлены хромосомной патологией и, как правило, представлены единственными случаями в семье, а для других описаны альтерации одного или нескольких генов с различным типом наследования.


Хромосомная патология часто сочетается с ярко выраженными фенотипическими особенностями, во многом зависящими от протяженности локуса хромосомного дисбаланса,— грубой задержкой психомоторного (ПМР) и (или) речевого развития (РР), множественными пороками и стигмами дизэмбриогенеза, в связи с чем диагноз можно заподозрить уже в неонатальном периоде.
При генных альтерациях фенотип больных может быть крайне специфичен, благодаря чему зачастую осмотра пациента достаточно для постановки клинического диагноза, а генетическая верификация необходима для дифференциальной диагностики и последующего планирования деторождения.
В то время как ряд генетических синдромов можно заподозрить по клиническим и фенотипическим проявлениям (см. табл.), при большинстве наследственных онкологических заболеваний только ДНК-диагностика, анализ родословной и длительное наблюдение позволяют поставить диагноз. К числу подобных заболеваний относят синдромы Ли — Фраумени I типа (OMIM#151623, ген TP53) и II типа (OMIM#609265, ген CHEK2), фон Хиппеля — Линдау (OMIM 193300, ген VHL), предрасположенности к полиорганным опухолям — 2 (OMIM# 619975, ген MBD4), Линча (OMIM#120435, ген MSH2; OMIM#609310, ген MLH1; OMIM#614337, ген PMS2; OMIM#614350, ген MSH6), DICER1‑синдр ом (OMIM#138800 и OMIM#601200, ген DICER1), наследственные формы ретино- (OMIM#180200, ген RB1) и нефробластомы (OMIM#194070, ген WT1), а также ряд других синдромов.
В нашей практике неоднократно встречались пациенты с наследственными онкологическими синдромами. Приводим клиническое наблюдение пациента с синдромом Ли — Фраумени, для которого характерно развитие первично-множественных ЗНО различных локализаций (саркома, рак молочной железы, лейкоз, опухоль надпочечников и др.) с манифестацией в раннем возрасте, а также аутосомно-доминантный тип наследования с высокой пенетрантностью. Прогноз заболевания, как правило, неблагоприятный. Ввиду высокого риска развития ЗНО в течение всей жизни пациенты должны динамически проходить комплексное обследование, включая МРТ всего тела.
КЛИНИЧЕСКОЕ НАБЛЮДЕНИЕ
Больной, 10 лет, проходил лечение в НИИ ДОиГ имени академика Л.А. Дурнова с диагнозом «остеосаркома диафиза правой бедренной кости, манифестация в 5 лет 5 месяцев». Семейный анамнез отягощен ЗНО различных локализаций (рис. 1).
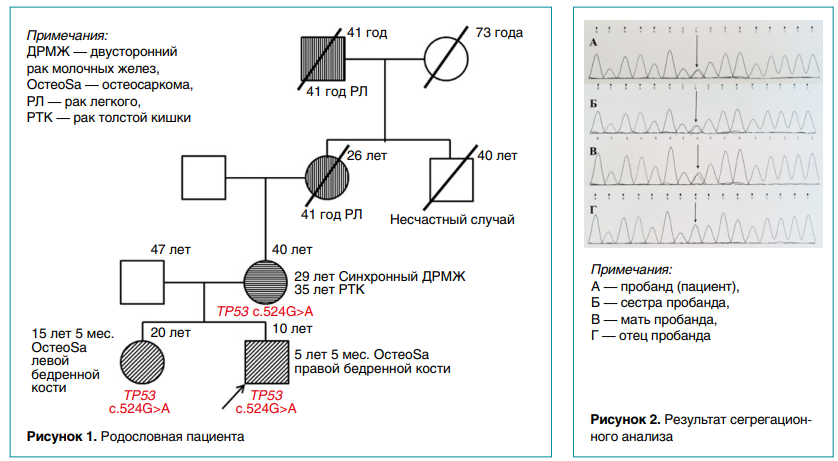
По данным молекулярно-генетического исследования, методом NGS (секвенирования следующего поколения) в 5‑м экзоне гена ТР53 выявлен герминальный патогенный вариант NM_000546.6(TP53): c.524G>A (p.Arg175His, rs28934578) в гетерозиготном состоянии. При выполнении сегрегационного анализа методом секвенирования по Сенгеру аналогичный вариант нуклеотидной последовательности был выявлен у матери и сестры пациента (рис. 2). На основании результатов проведенного исследования, а также семейного анамнеза, пациенту был поставлен диагноз «синдром Ли — Фраумени I типа, передача по материнской линии». Рекомендовано пожизненное динамическое наблюдение и планирование деторождения ввиду высокого риска (50 %) передачи мутации потомству. Диагностика наследственных онкологических синдромов у детей является принципиально важной для планирования дальнейшего наблюдения и лечения. Так, например, пациентам с синдромами Ли — Фраумени и Ниймегена следует ограничить использование лучевых методов исследования и отказаться от лучевой терапии ввиду высокого риска развития вторых опухолей. Своевременно поставленный генетический диагноз позволяет также избежать повторного рождения больных детей в семье при условии планирования беременности, выполнения пренатальной и предимплантационной диагностики. Для верификации наследственных онкологических синдромов у детей используются различные кастомные панели генов. Основной метод исследования — NGS, однако в ряде случаев для выявления протяженных внутригенных делеций может потребоваться мультиплексная лигазная цепная реакция (MLPA). В случае хромосомной патологии пациентам могут быть выполнены цитогенетическое исследование или хромосомный микроматричный анализ. Для решения вопроса о показаниях и объеме молекулярно-генетического исследования рекомендуется консультация генетика
НАШИ ПАРТНЕРЫ