Статьи
Врожденные дефекты иммунитета и их значение для эпидемиологии инфекционных и неинфекционных заболеваний
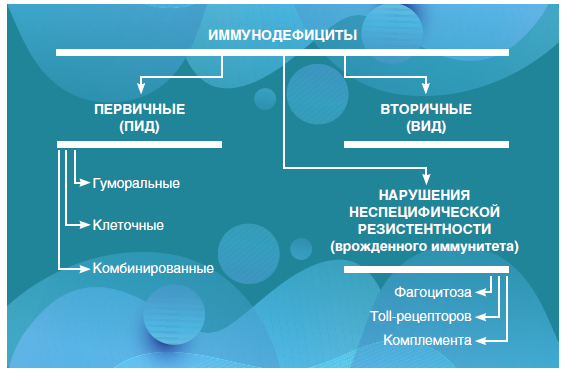
 Статья посвящена врожденным дефектам иммунитета или, более привычно, первичным иммунодефицитам (ПИД). В основе этой многообразной, активно расширяющейся группы состояний лежат генетически обусловленные дефекты иммунной системы. ПИД насчитывают более 450 заболеваний, подразделяющихся на 10 групп в зависимости от основного иммунологического патомеханизма.
Статья посвящена врожденным дефектам иммунитета или, более привычно, первичным иммунодефицитам (ПИД). В основе этой многообразной, активно расширяющейся группы состояний лежат генетически обусловленные дефекты иммунной системы. ПИД насчитывают более 450 заболеваний, подразделяющихся на 10 групп в зависимости от основного иммунологического патомеханизма.
ПОКАЗАТЕЛЬНЫЕ ПРИМЕРЫ
Основное проявление ПИД — инфекционный синдром, в связи с чем в середине прошлого века на первый план выходили ПИД с грубыми дефектами иммунитета, приводившими к рецидивирующим, тяжелым, нередко смертельным инфекциям, вызванным широко распространенными инфекционными возбудителями. Пациенты с дефектом продукции антител (классический пример — Х-сцепленная агаммаглобулинемия), как правило, страдают отитом, пневмонией и сепсисом, вызванными грамположительными или, реже, грамотрицательными бактериями.
По мере изучения ПИД были описаны целые группы состояний с более тонкими иммунологическими дефектами, обусловливающими особую чувствительность таких пациентов к редким и (или) оппортунистическим патогенам. В качестве примера можно привести хроническую гранулематозную болезнь с дефектом образования свободных радикалов кислорода в нейтрофилах и предрасположенностью к глубоким микозам, в первую очередь аспергиллезу.
Еще одним примером может служить группа ПИД с дефектами различных белков пути активации ϒ-интерферона и интерлейкина‑12. Для таких больных характерна особая чувствительность к микобактериозам — как типичному, вызванному M. tuberculosis, так и к системным инфекциям, спровоцированным нетуберкулезными микобактериями и вакцинальным штаммом M. bovis (БЦЖ). Последние данные говорят о том, что более 90 % пациентов с атипичными микобактериозами, включая БЦЖ-инфекцию, имеют генетически обусловленные дефекты иммунной системы.
Отдельная группа врожденных дефектов иммунитета — это пациенты с тяжелым и (или) атипичным течением широко распространенных или вновь возникающих вирусных инфекций. Так, в течение последнего десятилетия стало ясно, что больные с дефектами пути интерферонов первого типа могут оставаться совершенно здоровыми и не проявлять характерных для ПИД признаков в течение десятков лет до встречи с конкретным вирусом, способным приводить к смерти. Так бывает при респираторном дистресс-синдроме на фоне гриппа при наличии дефекта регуляторного фактора транскрипции интерферона‑7 (IRF7) или при инвалидизирующих последствиях герпетического энцефалита при дефекте толл-подобного рецептора‑3 (TLR3). Совсем свежий пример такой особой чувствительности — выявление первичных дефектов внутриклеточного иммунитета у 5 % пациентов с тяжелым или смертельным течением COVID‑19, а также вторичных (обусловленных аутоантителами) дефектов этого звена иммунитета у 20 % таких больных.
Дефекты некоторых иммунных механизмов предрасполагают к особому виду инфекционной чувствительности — неспособности иммунной системы пациентов контролировать онкогенный потенциал различных вирусов. У таких больных либо развиваются опухоли, характерные исключительно для иммунокомпрометированного хозяина (саркома Капоши, а также гладкомышечная опухоль, ассоциированная с вирусом Эпштейна — Барр [ВЭБ]), либо частота развития широко распространенных опухолей, например ВЭБ-ассоциированных лимфом, превышает среднепопуляционную в десятки раз, а прогноз терапии бывает крайне неблагоприятным. Знание подлежащего генетического и (или) иммунного дефекта в таких случаях позволяет выбрать адекватную тактику терапии опухолей — в какихто случаях более агрессивную, в том числе трансплантацию костного мозга, способную исправить дефект иммунной системы онкологического больного.
ЧЕМ ПОМОЖЕТ РЕГИСТР
Изучение различных групп иммунодефицитов и их патологических механизмов позволяет прогнозировать развитие тех или иных редких и (или) оппортунистических инфекционных и инфекционноссоциированных заболеваний у различных групп населения, в том числе с учетом территориальных особенностей. Описание распространенности видов ПИД в различных регионах России с помощью созданного Российского регистра первичных иммунодефицитов позволяет успешно осуществлять эту задачу.
Особенно важно, что полученная при анализе регистра информация помогает осуществлять профилактику редких и (или) оппортунистических инфекций путем раннего выявления пациентов с ПИД и проводить патогенетическое лечение, включая иммуномодулирующую и заместительную терапию препаратами иммуноглобулина человеческого нормального, а также радикальное лечение методом трансплантации гемопоэтических стволовых клеток.
Еще один важный эпидемиологический аспект изучения ПИД связан с тем, что пациенты с врожденными дефектами иммунитета нередко не в состоянии элиминировать инфекционные патогены в течение длительного времени, а значит, являются резервуаром и источником распространения различных инфекций.
Многочисленные исследования, включая наши собственные, говорят о носительстве пациентами с ПИД вакцинального и дикого штаммов полиомиелита, что служит одним из препятствий к его полной эрадикации в мире. Последние дан ные демонстрируют склонность больных ПИД к длительному (2–14 месяцев) бессимптомному или малосимптомному носительству вируса COVID‑19. Вследствие чего создается не только опасность инфицирования контактных лиц, но и риск мутирования вируса с появлением новых штаммов COVID‑19.
Таким образом, изучение различных аспектов распространенности, диагностики и терапии первичных иммунодефицитов имеет большое значение не только для теоретической и клинической иммунологии, но и для эпидемиологии инфекционных и инфекционно-ассоциированных заболеваний, и открывает новые перспективы снижения их частоты, а также профилактики осложнений.
Список литературы находится в редакции
НАШИ ПАРТНЕРЫ